METODOLOGÍA
En el experimento, vamos a analizar datos de secuenciación de genoma completo (shotgun) generados a partir de microorganismos presentes en muestras de vino. Las muestras de vino se han tomado en tres puntos distintos del proceso de vinificación: entrada en uva (mosto), fermentación alcohólica y fermentación maloláctica:
- EU_TJT19: ENTRADA EN UVA
- FA_TJT19: FERMENTACIÓN ALCOHÓLICA
- FM_TJT19: FERMENTACIÓN MALOLÁCTICA
A partir de estas muestras se ha aisló el ADN, se sintetizaron las librerías para la secuenciación, y se secuenciaron en el Servicio de Secuenciación Genómica del Instituto de Biología Funcional y Genómica utilizando una plataforma de secuenciación masiva Illumina NextSeq500, con una Flow Cell Mid-150 utilizando el protocolo paired-end.
Para explicar la metodologia, esta se basa resumidamente en un análisis metagenómico y el uso de programa MetaPhlan 3.0. es una herramienta para analizar la composición de las comunidades microbianas a partir de datos de secuenciación de genoma completo (shotgun sequencing data).
A parte del uso del programa methaplan tuvimos que realizar una serie de comandos en la terminal de linux que se muestran a continuación, explicando estos de uno en uno.
- En primer lugar se ejecuta el comando de methaplan para obtener de los datos de nuestro estudio una tabla con la composición microbiana de los distintos vinos a estudio.
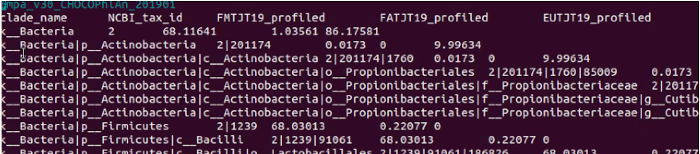
- Tras realizar este comando para cada una de las muestras se nos crea un archivo TSV con los microorganismos presentes en el vino en cada uno de los momentos antes mencionados de la vinificación
- Una vez tenemos las tablas para las distintas muestras, las unimos todas en una sola tabla que se llamará 'TJT19_merged_abundance_table.tsv', esto se realiza con el comando:
- Tras esto visualizamos que este archivo se ha creado con ls:
- En el paso 5 se visualizará la tabla con todas las muestras, y vemos que no vamos a necesitar todas las columnas de esta tabla para nuestro estudio.
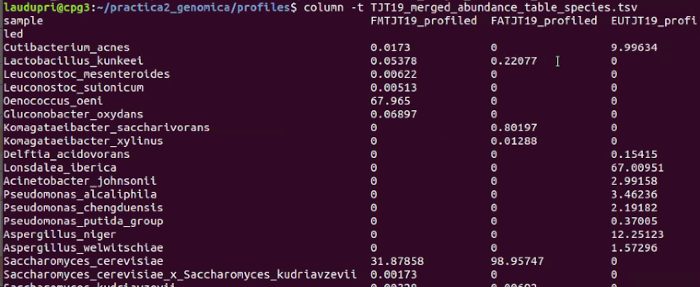
- Dado que no vamos a necesitar todas las columnas, con estos comandos nos quedamos solo con las columnas de la tabla que nos iteresen, y finalmente tendremos la tabla siguiente.